1119 Weekend Drug Holiday of Dasatinib in CML Patients Not Tolerating Standard
Dosing Regimens. Reducing Toxicity with Maintained Disease Control
Oral and Poster Abstracts
Poster Session: Chronic Myeloid Leukemia - Therapy Poster I
Saturday, December 5, 2009, 5:30 PM-7:30 PM
Hall E (Ernest N. Morial Convention Center)
Poster Board I-141
Paul La Rosee, MD1*, Armin Leitner, MD2*, Philippe Martiat, MD3, Thomas Klag,
MD1*, Samina Shazi, MD2*, Anne Treschl, MD2*, Martin C M黮ler, MD2*, Thomas
Schenk, MD1*, Benjamin Hanfstein2* and Andreas Hochhaus, MD1
1Hematology/Oncology, Universit鋞sklinikum Jena, Jena, Germany
2III. Medizinische Klinik, Universit鋞smedizin Mannheim, Universit鋞 Heidelberg,
Mannheim, Germany
3Dept. of Experimental Hem., Institut Jules Bordet, Brussels, Belgium
Dasatinib (DA) is a multitargeted tyrosine kinase inhibitor (TKI) approved for
2nd line treatment of chronic myelogenous leukemia (CML) patients after imatinib
failure. DA-related toxicity mandates dose reduction in selected patients beyond
the labelled reduced continuous dosing. In chronic phase (CP) patients,
intermittent targeting of BCR-ABL by a once daily regimen reduces side effects
with equal efficacy compared to the initially explored twice daily regimen.
Thus, considering the short half-life of DA (3-5 hours) additional treatment
interruptions to reduce the total weekly dose may not negatively affect
treatment outcome while allowing continued treatment with an effective drug. In
a retrospective analysis 33 CML patients (pts; 20 m, 13 f, median age 66 years,
range 39-81) with intolerance (n=11) or resistance (n=22) to imatinib were
investigated. Pts were selected based on the toxicity-guided administration of a
dose reduced dasatinib regimen and were treated with an on/off regimen (3 to 5
days on, 4 to 2 days off) expecting a reduction of DA dependent off-target
toxicity. Pts were followed by routine hematologic and cytogenetic assessment,
and molecular monitoring (quantitative reverse transcriptase polymerase chain
reaction, PCR) to safeguard clinical response to the altered drug schedule.
Further, resistant pts were regularly screened for BCR-ABL mutations. Median
time since CML diagnosis until start of DA treatment was 38 mo (range, 6-189).
The median number of preceding treatment modalities was 3 (range, 1-5). The
median follow up of interval treatment was 23 mo (range, 3-41). 30 patients were
in CP, 2 in accelerated phase, and one in blast phase CML. 13/33 patients
carried mutant BCR-ABL prior to onset of DA-treatment. Non-exclusive reasons for
dose reduction were hematologic toxicity (17/33; 51%), and fluid retention
(18/33, 55%), including 17 patients with pleural effusions. 27 patients (82%)
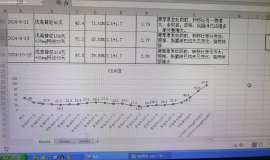
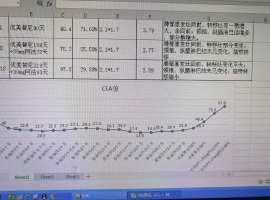
suffered from grade III/IV (CTC) side effects. The median weekly dose of the DA
weekend holiday schedule was 500mg (range, 320-500). During interval treatment,
mean CTC grade for hematologic toxicity improved from grade 3.2 to 1.5
(p<0.001), and for fluid retention from grade 2.9 to 1.6 (p<0.001). All but 2
pts (89%) affected by fluid retention, and all but one patient suffering from
hematologic toxicity (94%) achieved a lower CTC toxicity level by allowing drug
holiday. In 6/33 pts, resistance mutations (T315I x 2, F317L x 3, L248V) were
recovered. For response analysis, 2 pts were excluded due to early stem cell
transplantation or loss of follow up. 13/31 (42%) did either show transient
improved molecular response or remained on stable BCR-ABL load over time. 3/31
progressed to advanced phase CML. 18/31 (58%) pts showed the desired disease
control according to established criteria despite reduced total weekly DA doses
either demonstrated by achieving an improved response level (12/31), or keeping
the response level achieved by continuous dosing (6/31). Fourteen of the 18 pts
achieved or maintained major molecular response (MMR) with 5 pts repeatedly
tested negative by PCR. The remainder four pts demonstrated response by
achieving complete cytogenetic remission (CCyR, 2x) or reduced BCR-ABL load <1%
according to the international scale. Of note, 10/12 pts with improved response
have been treated for a minimum of 6 mo with continuous dosing DA regimens
without having achieved the response level observed after allowing drug holiday.
We conclude that weekend treatment interruption allows continuation of DA
treatment for pts suffering from side effects. This retrospective analysis in
pts resistant or intolerant to imatinib with up to 5 preceding treatment
modalities suggests good and in many cases even improved efficacy of interval
treatment compared to continuous dosing. These data mandate the initiation of
clinical trials to investigate alternative intermittent targeting regimens. |






 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡

 rescription Assistance Programs
rescription Assistance Programs